SAMlab - Pracownia analizy struktury, własności i zastosowań monowarstw SAM

W laboratoriach SAMlab zajmujemy się analizą nanostruktur organicznych wytwarzanych na drodze samoorganizacji (Self-Assembled Monolayers - SAM). Prowadzimy badania mające na celu optymalizację struktury i własności takich układów głównie pod kątem ich zastosowania w elektronice molekularnej i organicznej. W swojej analizie wykorzystujemy bardzo szeroką gamę technik mikroskopowych (STM/AFM), spektroskopowych (IRRAS, XPS, TP-XPS, NEXAFS), spektrometrycznych (SIMS, TP-SIMS) oraz pomiary przewodnictwa (eGaIn, C-AFM).
Dlaczego SAMy i ich optymalizacja są ważne?
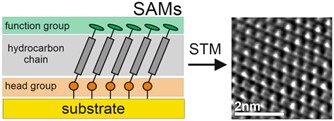
Łączenie materiałów nieorganicznych i organicznych staje się koniecznym dla dalszego rozwoju obecnych technologii w tak zróżnicowanych obszarach jak elektronika, biotechnologia czy nowe źródła energii. W każdym z tych zastosowań połączenie to prowadzi do powstania nieorganiczno-organicznego interfejsu, który dla zastosowań bazujących na nanotechnologii staje się jednym z najbardziej krytycznych elementów danego urządzenia. Ta kluczowa rola wynika z głębokich fizycznych i chemicznych różnic pomiędzy materiałami organicznymi i nieorganicznymi, co sprawia, że kontrola właściwości na ich styku jest szczególnie trudna. Samoorganizujące się monowarstwy (SAM) zapewniają prosty i niezawodny sposób kontrolowania granicy faz organicznych i nieorganicznych poprzez tworzenie predefiniowanej, ultra-cienkiej, jednocząsteczkowej monowarstwy organicznej spontanicznie chemisorbowanej na podłożach nieorganicznych. Struktura i właściwości SAM są wynikiem skomplikowanego wzajemnego oddziaływania trzech podstawowych elementów budujących te struktury tj. grupy funkcyjnej (functional group) grupy czołowej (head group) oraz łączącego te dwie grupy łańcucha węglowodorowego (Rys. 1). Niezależnie od konkretnego zastosowania SAM, najważniejszą cechą tych nanostruktur organicznych jest ich stabilność i właściwości przenoszenia ładunku. Znaczenie transferu ładunku jest raczej oczywiste, biorąc pod uwagę zastosowania w takich obszarach jak elektronika czy fotowoltaika, a interesujące są tutaj zarówno wysoce przewodzące, jak i wysoce izolujące układy SAM. Natomiast stabilność monowarstwy jest w rzeczywistości właściwością znacznie bardziej zróżnicowaną. W zastosowaniach w obrębie elektroniki czy fotowoltaiki stabilność termiczna ma kluczowe znaczenie biorąc pod uwagę zarówno podwyższoną temperaturę podczas formowania danego urządzenia (np. osadzania organicznych półprzewodników na powierzchni SAM) jak i wydzielanie ciepła podczas pracy całego układu. Natomiast w zastosowaniach związanych z biotechnologią stabilność termiczna jest zwykle mało istotna biorąc pod uwagę działanie w warunkach RT, jednakże stabilność chemiczna nabiera fundamentalnego znaczenia biorąc pod uwagę konkurencję SAM z innymi adsorbatami obecnymi zwykle w roztworach biologicznych. Ponadto dla wszystkich tych rozległych obszarów zastosowań litografia SAM-ów ma kluczowe znaczenie dla przestrzennego sterowania strukturą i właściwościami organiczno-nieorganicznego interfejsu, dlatego też istotny staje się jeszcze jeden ważny aspekt stabilności SAM, tj. stabilność wobec promieniowania jonizującego, w tym szczególnie stabilność względem wiązki elektronów, które zapewniają zarówno najwyższą możliwą rozdzielczość przestrzenną jak i pozwalają na przekształcanie SAM w inne materiały dwuwymiarowe (2D) takie jak grafen lub nanomembrany węglowe (CNM).
Przykładowe projekty badawcze
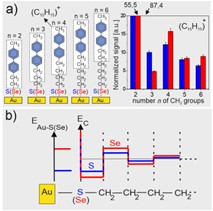
Analiza stabilności interfejsu molekuła-metal poprzez wykorzystanie techniki SIMS. W ramach tego projektu wykazaliśmy możliwość zastosowania techniki spektrometrii masowej jonów wtórnych (SIMS) w połączeniu z obliczeniami teorii funkcjonału gęstości (DFT) oraz symulacjami dynamiki molekularnej (MD) do analizowania stabilności wiązań chemicznych na interfejsie molekuła-metal. Badania przeprowadzone z wykorzystaniem modelowych układów SAM w postaci BPnS(Se)/Au(Ag) (CH3-C6H5-C6H5-(CH2)n-S(Se)/Au(Ag), których strukturę analizowaliśmy w ramach wcześniejszych badań podsumowanych ostatnio w pracy [1]. Uzyskane wyniki po raz pierwszy ujawniły istnienie efektu oscylacji stabilności kolejnych wiązań chemicznych w pobliżu interfejsu molekuła metal wynikających ze złamania symetrii translacyjnej w łańcuchu molekularnym (Rys. 2). [2] W badaniach pokazaliśmy, że amplituda tego zjawiska zależy zarówno od zastosowanej grupy wiążącej SAM do metalu (S/Se) jak i od wyboru podłoża metalicznego (Au/Ag) [3]. W trakcie realizacji tego projektu pokazano także możliwość wykorzystania analizy SIMS w funkcji zmian temperatury próbki, co prowadzi do analizy stabilności termicznej układu. Przeprowadzone badania ujawniły, że stabilność termiczna i chemiczna SAM mogą być przeciwnie skorelowane z uwagi na efekt oscylacji stabilności wiązań chemicznych. W tym przypadku oznacza to, że zwiększona energia wiązania Se-Au(Ag) w stosunku do S-Au(Ag) jest osiągana kosztem zmniejszenia energii kolejnego wiązania tj. Se-C w stosunku do S-C. O ile pierwsze z tych wiązań decyduje o stabilności chemicznej układu (np. w eksperymentach wymiany innymi molekułami) o tyle kolejne wiązanie jest najsłabszym w układzie i określa zakres jego stabilności termicznej.[4]
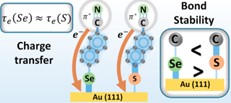
Wpływ struktury i stabilności samoorganizujących się monowarstw organicznych na proces ich przewodnictwa elektrycznego. Głównym osiągnięciem tego projektu było wykazanie korelacji pomiędzy stabilnością wiązania chemicznego na interfejsie molekuła-metal a przewodnictwem elektrycznym monowarstwy. Pomiary zostały przeprowadzone dla specjalnie zaprojektowanego układu bliźniaczych warstw SAM na bazie azobenzenu wiążących się z powierzchnią poprzez atom S lub Se oraz zakończonych grupą nitrylową mającą w tym eksperymencie podwójne znaczenie tj. umożliwiającą przeprowadzenie pełnej analizy orientacji molekuł na powierzchni (poprzez pomiary NEXAFS dla sygnału C oraz N) oraz przeprowadzenie pomiarów transferu ładunku poprzez zastosowanie techniki rezonansowej spektroskopii Auger’a (RAES) (Rys. 3). Pomiary spektroskopowe (IRRAS, XPS, NEXAFS) oraz mikroskopowe (STM) pokazały [5], że oba typy monowarstw tworzą bardzo podobne struktury, co dawało podstawę do przeprowadzenia wiarygodnej analizy stabilności wiązania tych monowarstw z powierzchnią [5], stabilności termicznej [4] oraz analizy transferu ładunku [5]. Pomiary stabilności oparte na wymianie molekuł ujawniły większą stabilność wiązania Se-Au w porównaniu do S-Au [2]. Kluczowe okazały się jednak pomiary SIMS [2], które nie tylko potwierdziły tą zależność ale przede wszystkim pokazały, że zwiększenie energii wiązania Au-Se odbywa się kosztem zmniejszenia energii sąsiedniego wiązania Se-C w porównaniu z układem opartym na siarce. Ta obserwacja pozwoliła na wytłumaczenie zaskakującego wyniku pomiaru czasu transferu ładunku przez układ, który jak się okazało nie zależy od siły wiązania molekuły do podłoża. Co ważne wynik ten jest sprzeczny z przewidywaniami, jakie sformułowano we wszystkich wcześniejszych pracach analizujących ten podstawowy dla elektroniki molekularnej problem. Brak takiej zależności wynika naszym zdaniem z tego, że zwiększenie energii wiązania do podłoża (Se-Au > S-Au) odbywa się za cenę zmniejszenia energii sąsiedniego wiązania chemicznego (Se-C < S-C), co w efekcie prowadzi jedynie do redystrybucji gęstości elektronów walencyjnych na interfejsie molekuła-metal i nie wpływa na efektywną zmianę czasu tunelowania przez układ. Ponadto unikalne połączenie wyników analizy stabilności (SIMS) w tych wzorcowych układach z obliczeniami teoretycznymi (DFT), jakie zostały dla nich wykonane, pozwoliło na zaproponowanie geometrii wiązania molekuł na bazie tioli i selenoli do podłoża Au(111) poprzez dodatkowe atomy podłoża (adatoms) [4]. W projekcie tym analizowaliśmy także formowanie monowarstw SAM na bazie alkynów (CH3-(CH2)n-CC-Au) w których grupą wiążącą do powierzchni metalu jest węgiel. Nasze badania ujawniły po raz pierwszy, że związki te formują wysoce uporządkowane monowarstwy, których struktura jest bardzo zbliżona do tej jaką znamy dla klasycznych monowarstw SAM opartych na alkanotiolach.[6] Pomiary przewodnictwa pokazały, że pomimo znacznie większej stabilności wiązania Au-C w porównaniu do Au-S przewodnictwo monowarstw opartych na alkynach jest w granicach dokładności podobne do monowarstw opartych na tiolach i to nie zależnie od tego czy analizujemy związki alifatyczne czy aromatyczne.[7,8] W projekcie tym analizowano także biologicznie inspirowane monowarswy SAM oparte na bazie peptydów oraz glikolu etylenowego ujawniając zaskakująco wysokie przewodnictwo elektryczne tych układów, co wskazuje na możliwość ich wykorzystania w elektronice organicznej.[9,10]
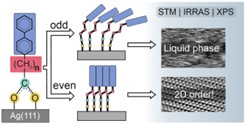
SAMy oparte na karboksylowej grupie wiążącej. W tym projekcie analizowaliśmy (STM/IRRAS/XPS) możliwość wykorzystania karboksylowej grupy wiążącej do formowania monowarstw SAM i ich zastosowania w elektronice molekularnej oraz do formowania materiałów 2D poprzez oddziaływanie z wiązką elektronową. W pierwszym etapie projektu poprzez porównanie analogicznych chemicznie i strukturalnie monowarstw BP2COO/Ag (CH3-C6H4-C6H4-(CH2)2-COO/Ag) and BP2S/Ag (C6H4-C6H4-(CH2)2-S/Ag) pokazaliśmy, że zamiana grupy wiążącej z tiolowej na karboksylową pozwala na około 30 krotne zwiększenie struktur domenowych a przez to analogiczny poziom redukcji defektów przy jednoczesnej 300 krotnej redukcji czasu formowania monowarstwy.[11] Analiza serii homologicznej BP2COO/Ag (n = 1-6) ujawniła bardzo silny efekt parzystości występujący w tych monowarstwach, w których dla parzystych wartości parametru n formowane są krystalicznie uporządkowane struktury, a więc odpowiednie do zastosowań elektroniki molekularnej wymagającej jak najmniejszej koncentracji defektów, natomiast dla wartości nieparzystych powstaje nieuporządkowana („ciekła”) struktura odpowiednia do zastosowania w zakresie biotechnologii, gdzie mononowarstwa SAM powinna móc dopasować się do amorficznych struktur biologicznych (Rys. 4). [12] Nasze badania pokazały, że ten strukturalny efekt parzystości ma bezpośredni wpływ na wydajność i przebieg procesu modyfikacji tych monowarstw poprzez naświetlanie niskoenergetyczną (50 eV) wiązką elektronową [13] oraz że dla odpowiednio dobranych monowarstw proces ten prowadzi do formowania nanomembran węglowych (CNMs), które są wolne od zanieczyszczeń siarki.[14] W kolejnym etapie tego projektu analizowaliśmy wpływ zmiany grupy wiążące (z S lub Se na COO) przewodnictwo elektryczne pokazując, że zmiana ta nie ma istotnego znaczenia oraz że w przypadku zastosowania monowarstw bazujących na naftalenie można osiągnąć wyjątkowo wysoko przewodzące monowarstwy SAM o dużej stabilności termicznej, a więc bardzo dobrze nadające się do zastosowań elektroniki organicznej.[15]
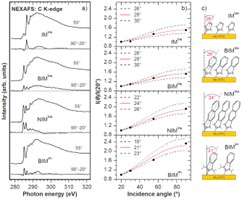
SAMy oparte na wykorzystaniu karbenów. W tym projekcie analizowaliśmy głównie możliwość formowania i wykorzystania monowarstw SAM na bazie N-heterocyklicznych karbenów (NHC) osadzanych na powierzchni złota poprzez utworzenie wiązania C-Au. Nasze badania ujawniły, że wbrew wcześniejszym analizom i przewidywaniom można formować wertykalnie zorientowane monowarstwy z wykorzystaniem molekuł NHC, w których grupy boczne atomów azotu tworzących strukturę NHC mają niewielkie rozmiary (Rys. 5). Zmniejszenie wielkości grup bocznych w oczywisty sposób wpływa na powierzchnię zajmowaną przez molekuły NHC w monowarstwie, a w ten sposób na ich gęstość upakowania. Przeprowadzona przez nas kompleksowa analiza spektroskopowa (XPS/NEXAFS) z wykorzystaniem serii molekuł NHC wykazała, że w przypadku formowania monowarstw z roztworu, taka modyfikacja struktury molekuł NHC pozwala zachować ich wertykalną orientację prowadząc do podwojenia gęstości upakowania w stosunku do typowych monowarstw NHC [16,17].
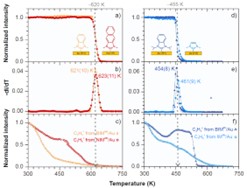
Badania te ujawniły także (Rys. 6), że odpowiednie zaprojektowanie struktury molekuł NHC poprzez wykorzystanie niewielkich grup bocznych oraz odpowiednią strukturę rdzenia molekuły umożliwia stworzenie monowarstw SAM na powierzchni złota o rekordowo dużej stabilności termicznej (energia desorpcji 1.9 eV).[17] Ponadto biorąc pod uwagę, że molekuły tworzące takie ultra-stabilne monowarstwy są zakończone pierścieniem aromatycznym lub naftalenem możliwe jest ich zastosowanie w zakresie elektroniki molekularnej. Ten zakres zastosowań jest nie tylko interesujący z uwagi na wysoką stabilność termiczną układów NHC ale także z uwagi na ich własności przewodnictwa. Nasze badania wskazują na to, że przewodnictwo tych monowarstw jest o około 4 rzędy wielkości niższe niż molekuł alkanotioli, które są uważane za silnie izolujące.[16] Oznacza to, że monwarstwy te mogą stanowić bardzo ciekawą alternatywę dla (i) formowania ultra-cienkich warstw izolujących organiczny półprzewodnik od elektrody bramki w tranzystorach typu OFET lub (ii) w przypadku zastosowania dłuższego rdzenia aromatycznego stanowić aktywny element tranzystora typu SAMFET tj. układu w którym monowarstwa SAM ma niskie przewodnictwo w kierunku prostopadłym do podłoża metalicznego, na którym jest formowana (zapewniając izolację od elektrody bramki) i wysokie przewodnictwo w kierunku równoległym do podłoża (zapewniając wydajny transport wzdłuż kanału łączącego elektrody źródła i drenu).
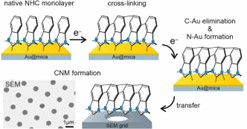
W tym projekcie po raz pierwszy analizujemy także naświetlanie elektronowe NHC SAM, wykorzystując w tym celu szereg monowarstw o różnej liczbie ugrupowań benzenowych i różnej wielkości bocznych grup azotowych, aby zmodyfikować ich gęstość upakowania oraz stabilność chemiczną (Rys. 7).[18] Zmiany grubości i składu monowarstwy były analizowane (XPS) w funkcji dawki elektronów. Nasze wyniki dostarczają prostych zasad projektowania optymalizujących strukturę NHC SAM pod kątem ich skutecznej modyfikacji przez wiązkę elektronową. Taka optymalizacja staje się szczególnie interesująca, biorąc pod uwagę, że analizowane monowarstwy NHC wykazują nawet 100-krotnie wyższą stabilność swojego wiązania z metalowym podłożem wobec napromieniania elektronami w porównaniu ze standardowymi tiolami czy kwasami karboksylowymi. Zatem monowarstwy NHC oferują nową i ekscytującą alternatywę dla litografii chemicznej, w której należy ograniczyć modyfikację strukturalną SAM za pomocą wiązek elektronów lub fotonów (uszkodzenia SAM wywołane promieniowaniem rentgenowskim, UV lub EUV są w rzeczywistości spowodowane fotoelektronami i elektronami wtórnymi) głównie do grupy końcowej, zachowując nienaruszone wiązanie z podłożem. Ponadto wykazujemy pomyślną delaminację i przenoszenie napromieniowanych elektronami monowarstw NHC, co pozwala na tworzenie nanomembran węglowych (CNM). Jak pokazujemy, proces ten jest również bardzo wrażliwy na strukturę NHC SAM i przy odpowiednio zaprojektowanym systemie umożliwia tworzenie ciągłych, wolnostojących CNM, które nie zawierają siarki, a zatem są znacznie bardziej odpowiednie do niektórych zastosowań ultra-filtracyjnych w porównaniu ze standardowymi CNM wytwarzanymi z tioli, które są zanieczyszczone reaktywną siarką.
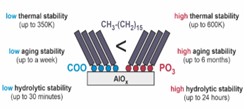
Formowanie monowarstw SAM na powierzchni naturalnego tlenku aluminium. Ten projekt dotyczy optymalizacji struktury monowarstw SAM tworzonych na znacznie bardziej dostępnym i powszechnie stosowanym w zakresie elektroniki i medycyny materiale jakim jest aluminium. Nasze badania w tym projekcie są głównie inspirowane zastosowaniem SAM jako pokrycia elektrody bramki w organicznych tranzystorach polowych (OFET), którą stanowi utlenione powierzchniowo aluminium. Wydajność tych urządzeń opiera się na stopniu krystaliczności półprzewodnika organicznego (OSC) w obszarze kanału przewodnictwa (zwiększona mobilność nośników ładunku), jakości izolatora i właściwym rozkładzie ładunku na granicy faz bramka-OSC (zmniejszone prądy upływowe na izolatorze). Poprawa wszystkich tych parametrów wymaga utworzenia wysokiej jakości dobrze zdefiniowanych karboksylowych SAM o najniższej możliwej koncentracji defektów. Badając kluczowe parametry wytwarzania SAM, takie jak rodzaj rozpuszczalnika, czas inkubacji i temperatura inkubacji oraz stosując kombinację kilku uzupełniających się technik eksperymentalnych (NEXAFS, XPS, IRRAS, AFM), zaproponowaliśmy zestaw parametrów umożliwiając otrzymanie gęsto upakowanych i dobrze uporządkowanych alifatycznych (izolujących) SAM z karboksylową grupą czołową na naturalnie utlenionym aluminium (Fig. 8).[19] Nasza analiza strukturalna, pokazała, że wbrew obiegowej opinii najwyższej jakości monowarstwy na bazie karboksyli uzyskuje się stosując „złe” rozpuszczalniki i relatywnie niską temperaturę inkubacji, co można wytłumaczyć interkalacją cząsteczek rozpuszczalnika w matrycy SAM. Ponadto pokazaliśmy, że naturalnie utworzona warstwa wierzchnia AlOx (x ~ 1,38) o grubości ~ 0,8 nm tworzy warstwę dipolową, które podobnie jak dedykowane monowarstwy SAM umożliwia zmianę pracy wyjścia elektronów z metalu na którym jest utworzona. W kolejnym kroku przeprowadziliśmy analogiczne badania strukturalne dla monowarstw, w których zamieniliśmy grupę karboksylową na fosfonową. Nasza analiza pokazała, że jakość wytworzonych monowarstw dla właściwie dobranego rozpuszczalnika jest tak samo dobra jak w przypadku modelowych monowarstw tioli na powierzchni złota. Te badania pokazały także, że stabilność hydrolityczna strukturalnie zoptymalizowanych fosfonowych SAM na AlOx jest około 50 razy wyższy w porównaniu do analogicznych SAM na bazie karboksyli. Z kolei badania starzenia w warunkach laboratoryjnych wykazały wynoszącą do 6 miesięcy stabilność fosfonowych SAM na AlOx w porównaniu do jedynie tygodniowej stabilności analogicznych karboksyli. W tym projekcie pokazaliśmy również, że granica stabilności termicznej strukturalnie zoptymalizowanych fosfonowych SAM na AlOx przekracza stabilność analogicznych monowarstw karboksylowych o około 250 K, a standardowych monowarstw alkanotioli na złocie o około 200 K. Biorąc pod uwagę, znaczną dyssypacje ciepła w układach elektroniki organicznej jak również podwyższoną temperaturę w której są one wytwarzane (np. w trakcie depozycji półprzewodnika organicznego na metalową elektrodę), bardzo wysoka stabilność termiczna czyni fosfonowe monowarstwy SAM na AlOx znakomitymi kandydatami do zastosowania w elektronice organicznej.
Wybrane publikacje
[1] Cyganik et al. Nano Res. 2024, 17, 4231. [2] Ossowski et al. Angew. Chem. Int. Ed. 2015, 54, 1336. [3] Ossowski et al. J. Phys. Chem. C 2017, 121, 459. [4] Ossowski et al. J. Phys. Chem. C 2017, 121, 28031. [5] Ossowski et al. ACS Nano 2015, 9, 4508. [6] Żaba et al. J. Am. Chem. Soc. 2014, 136, 11918 [7] Bowers et al. ACS Nano 2015, 9, 1471. [8] Bowers et al. J. Phys. Chem. C 2016, 120, 11331. [9] Baghbanzadeh et al. Angew. Chem. Int. Ed. 2015,127, 14956. [10] Baghbanzadeh et al. J. Am. Chem. Soc. 2017, 139,7624. [11] Krzykawska et al. Chem. Comm. 2017, 53, 5748. [12] Krzykawska et al. J. Phys. Chem. C 2018, 122, 919. [13] Kruk et al. J. Phys. Chem. C 2021, 125, 9310. [14] Neumann et al. ACS Appl. Mater. Inter. 2019, 11, 31176. [15] Wróbel et al. Adv. Electron. Mater. 2021, 7, 2000947. [16] Krzykawska et al. ACS Nano 2020, 14, 6043. [17] Wróbel et al. Nano Today 2023, 53, 102024. [18] Cegiełka et al. J. Phys. Chem. Lett. 2024, 15, 8196. [19] Cegiełka et al. Appl. Surf. Sci. 2023, 636, 157798. [20] Cegiełka et al. Appl. Surf. Sci. 2024, 665, 160199.Współpracownicy
- Prof. Michael Zharnikov, Heidelberg University, Germany
- Prof. George Whitesides, Harvard University, USA
- Prof. Andrey Turchanin, Friedrich Schiller University Jena, Germany
- Prof. Mariusz Krawiec, Marie-Curie University, Lublin, Poland
- Prof. Egbert Zojer, TU Graz, Austria
Członkowie grupy
- prof. dr hab. Piotr Cyganik - Kierownik
- mgr Daria Cegiełka
- mgr Agnieszka Grabarek
- dr Mateusz Wróbel
- Małgorzata Lebica
- Mark Kasatkin
- Karolina Sorn
- Marta Szatny
Byli członkowie grupy
- dr Maciej Dendzik
- dr Joanna Dudek (Sobczuk)
- mgr Dominika Gnatek
- dr Monika Kruk (Szwed)
- dr Anna Krzykawska
- mgr Małgorzata Niemiec
- mgr Agnieszka Noworolska
- dr Jakub Ossowski
- mgr Katarzyna Szelągowska
- dr Agnieszka Szpak
- dr Tomasz Żaba
 prof. dr hab. Piotr Cyganik
prof. dr hab. Piotr Cyganik