Pracownia komputerowego modelowania powierzchni

Używamy symulacje komputerowe do badania procesów powierzchniowych stymulowanych uderzeniami energetycznych pocisków w układy 3- (3D) i 2-wymiarowe (2D). Interesuje nas badanie procesów fizykochemicznych prowadzących do emisji cząstek z cienkich warstw metalicznych i półprzewodnikowych, nanostruktur organicznych, a także z grafenu, oraz z jedno i wielościennych nanorurek węglowych. Do badań używamy symulacji komputerowych opartych o formalizm dynamiki molekularnej.
Modelowanie komputerowe metodą dynamiki molekularnej
Symulacje komputerowe metodą dynamiki molekularnej (MD) służą do modelowania i wizualizacji badanych zjawisk. Od zarania technologii komputerowej, symulacje komputerowe stały się ważnym partnerem dla pomiarów doświadczalnych. Jeśli obserwuje się jakieś nowe zjawisko, zwykle chcemy zrozumieć jakie procesy są za nie odpowiedzialne. Symulacje komputerowe są szczególnie przydatne do osiągnięcia tego celu, ponieważ pozwalają zbadać wiele właściwości, których nie można bezpośrednio zmierzyć. Na przykład pozwalają zaglądnąć do wnętrza badanego układu i dokonać wizualnej inspekcji ruchów atomów prowadzących do wystąpienia określonego zdarzenia lub zbioru wydarzeń. Możemy po prostu zobaczyć co tam się dzieje! W podejściu MD, czasowa ewolucja danego układu jest modelowana przez rozwiązywanie równań ruchu Newtona dla wszystkich atomów tworzących dany układ. Siły działające na atomy są opisane przez połączenie empirycznych potencjałów dwu i wielociałowych.
Obliczenia przygotowawcze wykonujemy na naszym serwerze obliczeniowym wyposażonym w 356 jednostek obliczeniowych. Właściwe obliczenia są wykonywane na komputerach należących do sieci superkomputerowej PLGrid.
Cele badań

Szybki postęp miniaturyzacji, jaki dokonał się w ostatnich latach, spowodował duże zainteresowanie badaniem zjawisk fizycznych i chemicznych zachodzących w urządzeniach o wymiarach niewiele większych od wymiarów pojedynczych atomów. Zainteresowanie to zaowocowało poszukiwaniem technik doświadczalnych, które np. pozwoliłyby mierzyć i obrazować przestrzennie skład chemiczny w tej skali. Przykładami technik, które pozwalają na realizację tego zamierzenia są spektrometria masowa wtórnych jonów (SIMS) i spektrometria masowa wtórnych cząstek neutralnych (SNMS). Techniki te wykorzystuje zjawisko bombardowania powierzchni strumieniem pocisków o energiach keV-owych w celu wyrzucenia do próżni cząstek tworzących te powierzchnie. Zjawisko to nosi nazwę „rozpylanie”. Wyemitowane cząstki są następnie analizowane przy pomocy spektrometrów masowych. Z otrzymanych widm masowych wnioskuje się o tym, co znajdowało się na powierzchni. W układach wyposażonych w skanowaną, zogniskowaną wiązkę jonową można dodatkowo określić przestrzenne położenie danego związku w objętości próbki. Techniki SIMS/SNMS są tak czułe, że potrafią znaleźć pojedynczy atom lub cząsteczkę ukrytą wśród miliardów innych cząstek.

SIMS i SNMS mają wiele praktycznych zastosowań. Są wykorzystywane przy projektowaniu i wytwarzaniu układów scalonych, które napędzają nasze urządzenia elektroniczne. Służą również do określania wieku struktur geologicznych, pomagają w wykrywaniu fałszerstw dzieł sztuki. Pomagają również w łapaniu przestępców na podstawie dowodów znalezionych na miejscu zbrodni. Jednak najbardziej fascynującym zastosowaniem technik SIMS/SNMS jest ich wykorzystanie w badaniach medycznych i diagnostyce. Na przykład techniki te mogą tworzyć trójwymiarowe, chemiczne obrazy komórek biologicznych. Dzięki temu odgrywają istotną rolę w opracowywaniu nowych leków. Kompaktowe i w pełni automatyczne systemy SIMS są wprowadzane do szpitalnych sal operacyjnych, gdzie wspomagają zabiegi chirurgiczne, poprzez szybką identyfikację złośliwych tkanek nowotworowych. Dzięki temu chirurg od razu wie czy dany narząd należy usunąć. Taka sama identyfikacja, wykonana w tradycyjny sposób, może trwać kilka dni. Jeżeli diagnoza jest niepomyślna, pacjent musi ponownie trafić na salę operacyjną.
Głównym celem naszych badań wyjaśnienie co dzieje się w próbkach podczas ich analizy. W tym celu modelujemy procesy prowadzące do emisji cząstek z materiałów bombardowanych strumieniem cząstek o energiach keV-owych. Badamy jak parametry wiązki rozpylającej powierzchnie (rodzaj pocisków, ich energie kinetyczne i kąty padania) wpływają na wydajność i inne charakterystyki procesu emisji. W szczególności interesuje nas badanie procesów fizykochemicznych występujących w nowych konfiguracjach analitycznych. Przykładem takich konfiguracji jest użycie chemicznie złożonych pocisków klastrowych, zastosowanie dwuwymiarowych lub płynnych podłoży. Badania nad płynnymi próbkami są szczególnie istotne, ponieważ woda jest głównym składnikiem naszego organizmu. Dlatego ważne jest, aby zrozumieć i wyjaśnić, co dzieje się w takim środowisku podczas analizy SIMS/SNMS. Do realizacji naszych badań używamy symulacji komputerowych.

Przykładowe projekty badawcze
Bombardowanie powierzchni pociskami klasterowymi
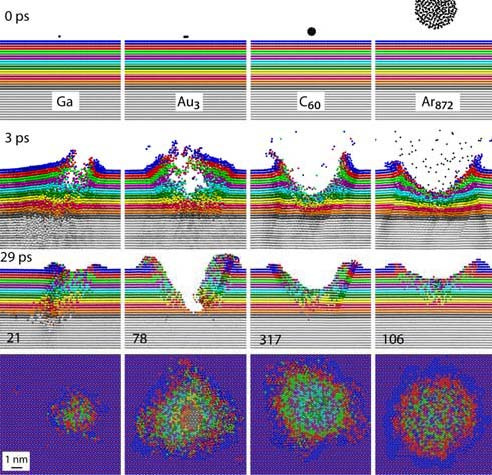
Pociski klastrowe pojawiły się w technikach SIMS/SNMS, gdy okazało się, że pozwalają na znaczne zwiększenie wydajności emisji cząstek, połączone z mniejszym prawdopodobieństwem fragmentacji emitowanych molekuł. Ponieważ w technikach SIMS/SNMS identyfikacja próbkowanego materiału odbywa się poprzez analizę widm masowych emitowanych cząstek, dlatego tak ważne jest, aby widmo to składało się głownie z oryginalnych molekuł, a nie ich fragmentów. To właśnie symulacje komputerowe pozwoliły zrozumieć i wyjaśnić dlaczego bombardowanie pociskami klasterowymi tak różni się od bombardowania monoatomowego. Uderzenie pocisku atomowego prowadzi do wytworzenia w materiale klasycznej kaskady zderzeń, w której pojedyncze atomy uderzają w inne atomy w sposób przypominający grę, w bilard. Jednak w tym przypadku cała energia niesiona jest przez jeden atom. Uderzenie takiego pocisku w molekułę prowadzi do jej rozbicia na kawałki. Pociski atomowe są niewielkie. Z łatwością przenikają do wnętrza materiału. W rezultacie ich energia jest deponowana głęboko w próbce, daleko od powierzchni. Tylko energia zdeponowana w pobliżu powierzchni próbki może być użyta do emisji cząstek. W rezultacie wydajność emisji jest w takich przypadkach niewielka. Energia zdeponowana głęboko powoduje jedynie mieszania materiału próbki oraz prowadzi do powstania uszkodzeń wewnątrz materiału. Obydwa te procesy zmieniają oryginalną strukturę geometryczną i skład chemiczny próbki, uniemożliwiając wykonanie analizy chemicznej z dużą przestrzenną zdolnością rozdzielczą [1].
Pociski klasterowe mają znacznie większy rozmiar niż pociski monoatomowe. W rezultacie, nie są one w stanie wniknąć do wnętrza materiału i deponują swoją energię blisko powierzchni. Skutkiem płytkiego deponowania energii jest wystąpienie silnej emisji cząstek z powierzchni materiału. Na przykład pocisk C60 powoduje emisję 15 razy większą niż pocisk Ga o tej samej energii kinetycznej. Emisji dużej liczby cząstek powoduje, że na bombardowanej powierzchni tworzy się krater, tak, jak to pokazano po lewej stronie. Ponieważ energia dzielona jest na wiele atomów w pocisku klasterowym, energia niesiona przez pojedynczy atom jest niewielka. W dodatku, ruch tych atomów jest przestrzennie skorelowany. W rezultacie, uderzenie pocisków klastrowych jest znacznie łagodniejsze od uderzenia ich odpowiedników atomowych. Skutkiem tego jest możliwość emisji z powierzchni nienaruszonych molekuł organicznych, czego nie można było osiągnąć pociskami monoatomowymi. Dlatego też, pociski klastrowe (np. C60, Arn) są obecnie stosowane do przeprowadzania przestrzennego obrazowania chemicznego struktur organicznych i biologicznych. Jednak ciągle poszukuje się nowych pocisków klasterowych, które pozwoliłyby na zwiększenie wydajności emisji. Ciekawymi, obecnie badanymi kandydatami są, np. pociski klasterowe zbudowane z molekuł H2O lub CO2.
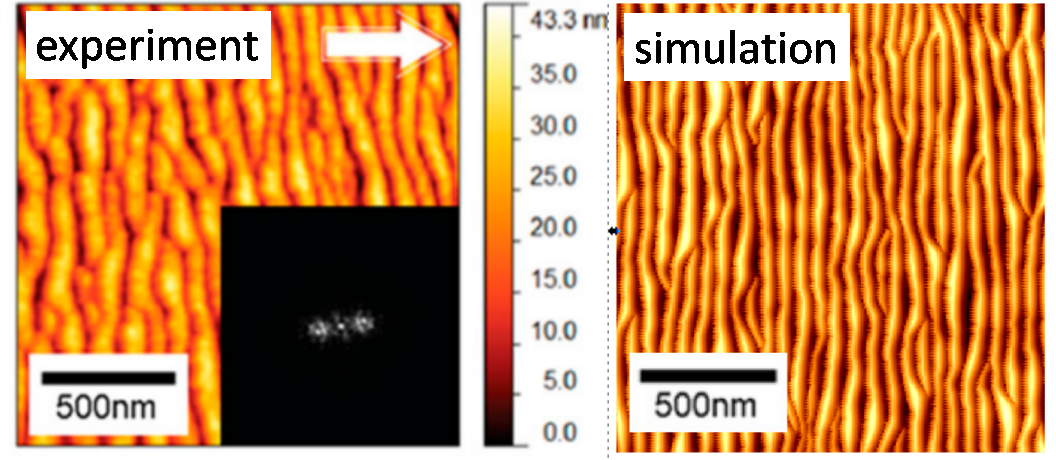
Jednym z najważniejszych czynników wpływających na przestrzenną dokładność pomiaru składu chemicznego, czyli tzw. rozdzielczości głębokościowej, jest pojawianie się podczas analizy chropowatości powierzchni. Jednym z rodzajów takiej chropowatości są zmarszczki (ripple). Pojawienie się zmarszczek jest czynnikiem ograniczającym możliwą do uzyskania głębokościową zdolność rozdzielczą, ponieważ dokładność ta nie może był większa niż amplituda zmarszczek. Niekiedy amplituda ta może być ogromna. Problem powstawania zmarszczek był szeroko badany w przypadku pocisków monoatomowych. Jednak niewiele było wiadomo na temat tego procesu w przypadku, gdy powierzchnia jest bombardowana przez pociski klasterowe. Niedawno zaproponowaliśmy prosty model, który pozwala na intuicyjne wyjaśnienie zjawiska tworzenia się zmarszczek poprzez transfer masy wywołanej uderzeniem pocisku klasterowego [2]. Kluczowy okazał się kształt zależności tego transferu od kąta padania wiązki. Łącząc ten model z symulacjami komputerowymi możemy przewidzieć kiedy pojawią się zmarszczki i co zrobić, aby ich uniknąć. Pomimo swojej prostoty model ten pozwala na uzyskanie wyników, które są ilościowo zgodne z wynikami pomiarów doświadczalnych. Wykorzystując ten model można pokazać, że problemy wynikające z pojawienia się zmarszczek można zmniejszyć poprzez zmniejszenie kąta padania wiązki rozpylającej w stosunku do normalnej powierzchni, lub poprzez zwiększenie jej energii kinetycznej.
Rozpylanie i modyfikacja układów dwuwymiarowych 2D (grafen, nanorurki weglowe)
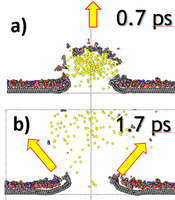
W naszych pracach skupiamy się na badaniu procesów zachodzących podczas analizy metodą SIMS/SNMS dwuwymiarowych (2D) materiałów. Układy te są ważne ze względu na ich niezwykłe optyczne, mechaniczne i elektronowe właściwości transportowe. Większość uwagi jest skupiona na grafenie, jako głównym materiale przyszłej elektroniki 2D. Jednak materiały takie jak: heksagonalny azotek boru (h-BN) i disiarczek molibdenu (h-MoS2) są również kluczowe, ponieważ mają podobne właściwości strukturalne jak grafen, ale różnią się zarówno przewodnością elektryczną, jak i właściwościami sprężystymi. Uzyskanie możliwości obrazowania struktury chemicznej dwuwymiarowych materiałów i ich interfejsów jest kluczowa dla tworzenia wysoce wydajnych ultracienkich urządzeń elektronicznych. Jedną z technik, w których pokłada się duże nadzieje jest SIMS/SNMS.
Nasze ostatnie badania pokazują również, że użycie podłoża grafenowego może zwiększyć czułość analizy SIMS o rzędy wielkości. W dodatku, zastosowanie podłoża grafenowego zwiększa prawdopodobieństwo emisji całych molekuł. Symulacje komputerowe pokazały, że materiał organiczny osadzony na grafenie jest wyrzucany w wyniku delikatnej interakcji z deformującym się podłożem grafenowym, tak jak to pokazano po lewej stronie. Te obserwacje potencjalnie otwierają drogę do wykonywania analizy chemicznej nanoobiektów, zespołów supramolekularnych oraz do detekcji substancji na poziomie attomolowym [3, 4].
Bombardowanie ciekłych matryc pociskami klasterowymi
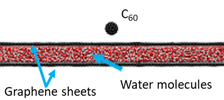
Chemiczne obrazowanie struktur biologicznych jest obecnie jednym z najgorętszych tematów badawczych opartych o wykorzystanie technik SIMS i SNMS. W zasadzie, najbardziej odpowiednim podłożem do wykonywania takich badań jest środowisko wodne, która jest naturalnym środowiskiem dla biopróbek. Jednak wykorzystanie matryc wodnych w SIMS-ie było do tej pory utrudnione przez problemy związane z niekompatybilnością warunków wysokiej próżni wymaganych do działania SIMS/SNMS, oraz próbek wodnych, powodowaną przez dużą prężność par tego środowiska. Jednym z ostatnio zaproponowanych sposobów przezwyciężenia tego ograniczenia było kapsułkowanie badanych biostruktur w cieczy zamkniętej między dwiema membranami grafenowymi, tak jak pokazano po lewej stronie. Grafen ma wystarczająco gęstą strukturę, aby zapobiec przedostawaniu się molekuł wody do próżni. Niestety, symulacje komputerowe pokazały, że po uderzeniu pocisku prowadzącym do powstania porów w grafenie, grafen ulega niemal natychmiastowemu „samoleczeniu”, co blokuje emisję badanej substancji z wnętrza kapsułki, uniemożliwiając wykonanie jej analizy [5].
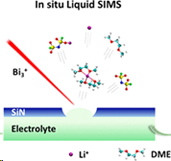
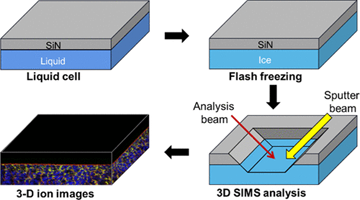
Inną możliwością badania ciekłych próbek w próżni jest zamknięcie badanej cieczy w małej komórce mikroprzepływowej. Komórka taka jest od góry zamknięta cienką membraną wykonana z SiN, tak jak to pokazano na rysunku po lewej stronie. Po wprowadzeniu tej komórki do komory analitycznej, zogniskowana wiązka jonów wierci w SiN mały otwór o średnicy około 2 μm. Otwór ten jest na tyle mały, że w komorze badawczej nadal można utrzymać wystarczająco wysoką próżnię (<5 × 10–7 mbar), która pozwala na pracę układu SIMS. System oparty o mikrokomórkę przepływową pozwolił już na uzyskanie wiele zagadkowych obserwacji, które wymagają teoretycznego wyjaśnienia [6]. Symulacje MD mogą w tym pomóc.
Opracowywanie szybkiego potencjału typu ReaxFF
Oprócz znacznego postępu jaki dokonał się w ostatnich latach w modelowaniu komputerowym zjawisk związanych z SIMS, nadal istnieje bardzo ograniczona liczba układów organicznych, które można modelować w pełnym atomistycznym podejściu. Sytuacja ta jest spowodowana koniecznością użycia znacznie bardziej wyrafinowanych potencjałów wielociałowych do opisu oddziaływania pomiędzy atomami w próbkach organicznych. Takie potencjały są bardzo złożone matematycznie, a przez to ich liczenie jest bardzo czasochłonne. Większość z potencjałów wieloatomowych, które można znaleźć w literaturze jest zbyt wolna, aby można je było zastosować w badaniach związanych z SIMS. W tych badaniach należy użyć próbek złożonych z milionów atomów, gdyż tylko taki układ jest w stanie odpowiednio pochłonąć pierwotną energię kinetyczną deponowaną przez typowy pocisk używany podczas analizy SIMS.
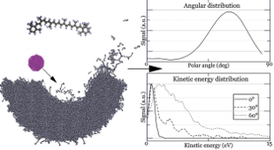
Część naszych prac jest poświęcona opracowaniu szybkiego potencjału do modelowania układów organicznych. W tym celu opracowaliśmy zmodyfikowaną wersję pola siłowego ReaxFF, która wyeliminowała czasochłonny proces równoważenia ładunku, zawiera rozszerzony zestaw treningowy dla faz skondensowanych i ma odpowiednią ścianę odpychającą potrzebną do modelowania zjawiska bombardowania cząstkami o energiach keV-owych. Do tej pory udało nam się opracować szybki potencjał pozwalający na opis oddziaływań w układach złożonych z atomów węgla i wodoru [7] oraz węgla wodoru i tlenu [8]. Przykład badań wykonanych z użyciem tego pola siłowego pokazano po lewej stronie. Celem badań było określenie wpływu kąta padania pocisku 10 keV C60 na efektywność emisji cząsteczek z warstw β-karotenu [9]. Badania pokazały, że całkowity sygnał emisji rośnie wraz z kątem padania, oraz, że najbardziej preferowanym kanałem wyrzutu jest emisja nienaruszonych cząsteczek. Przy okazji pokazano, że nowe pole siłowe pozwala na kilkakrotne przyspieszenie obliczeń bez utraty ich dokładności.
[1] B.J. Garrison and Z. Postawa, a chapter in "ToF-SIMS - Surface Analysis by Mass Spectrometry - 2nd Edition (2013).
[2] D. Maciazek, M. Kanski, and Z. Postawa, Analytical Chemistry 92 (2020) 7349.
[3] S. Verkhoturov, M. Gołuński, D. Verkhoturov, B. Czerwinski, M. Eller, S.Geng, Z. Postawa, E.A. Schweikert, J. Chem. Phys. 150 (2019) 160901.;
[4] M. Golunski, S. Hrabar, and Z. Postawa, Applied Surface Science 539 (2021) 148259
[5] S. Verkhoturov, M. Gołuński, Z. Postawa, w przygotowaniu.
[6] W. Guo, M. Kanski, W. Liu, M. Gołuński, Y. Zhou, Y. Wang, C. Cheng, Y. Du, Z. Postawa, W.D. Wei, and Z. Zhua, Analytical Chemistry 92 (2020) 13785 - wyróżniona publikacja.
[7] M. Kański, D. Maciazek, Z. Postawa, Ch.M. Ashraf, A.C.T. van Duin, B.J. Garrison, J. Phys. Chem. Lett. 9 (2018) 359.
[8] M. Kański, S. Hrabar, A.C.T. van Duin, Z. Postawa, J. Phys. Chem. Lett. 13 (2022) 628.
[9] M. Kański and Z. Postawa, Analytical Chemistry 91 (2019) 9161 - wyróżniona publikacja.
Członkowie grupy
- prof. dr hab. Zbigniew Postawa - Kierownik
- dr Michał Kański
- dr Soukaina Louerdi
- mgr Sviatoslav Hrabar
Okładki czasopism naukowych ilustrujące nasze badania
render(['images' => [ [ 'file_path' => '/img/groups/comp-sci/winter_2010.jpg', 'alt' => '', 'caption' => '' ], [ 'file_path' => '/img/groups/comp-sci/cover.jpg', 'alt' => '', 'caption' => '' ], [ 'file_path' => '/img/groups/comp-sci/cov150h_zp.jpg', 'alt' => '', 'caption' => '' ], [ 'file_path' => '/img/groups/comp-sci/cover_SIMS.jpg', 'alt' => '', 'caption' => '' ], [ 'file_path' => '/img/groups/comp-sci/cover_ancham.jpg', 'alt' => '', 'caption' => '' ], [ 'file_path' => '/img/groups/comp-sci/cover_anchem_2020.jpg', 'alt' => '', 'caption' => '' ] ]]); ?>Współpracownicy
- Gerhard Hobler, TU Wien, Austria
- Emile Schweikert, The Texas A&M University, USA
- Stanislav Verkhoturov, The Texas A&M University, USA
- Nicholas Winograd, The Pennsylvania State University, USA
Kontakt
 prof. dr hab. Zbigniew Postawa
prof. dr hab. Zbigniew PostawaWięcej informacji można znaleźć na stronie grupy: Strona grupy.